Ліпобон таблетки по 10 мг №30 (3 блістери х 10 таблеток)
- Все про товар
- Ціни в аптеках
- Відгуки1
")



Інструкція для Ліпобон таблетки по 10 мг №30 (3 блістери х 10 таблеток)
діюча речовина: езетиміб;
1 таблетка містить езетимібу 10 мг;
допоміжні речовини: целюлоза мікрокристалічна, манітол, натрію кроскармелоза, гідроксипропілцелюлоза низькозаміщена, повідон-К25, натрію лаурилсульфат, магнію стеарат.
Таблетки.
Основні фізико-хімічні властивості: круглі плоскі таблетки білого або майже білого кольору, з фаскою, з гравіюванням у вигляді стилізованої літери Е на одній і номера 612 на іншій стороні таблетки, без або майже без запаху.
Гіполіпідемічні засоби, монокомпонентні. Інші гіполіпідемічні засоби. Езетиміб. Код АТХ C10A X09.
Фармакодинаміка
Механізм дії
Езетиміб — це представник нового класу ліпідознижувальних речовин, які селективно пригнічують інтестинальну абсорбцію холестерину та відповідних рослинних стеролів. Езетиміб є перорально активним та має механізм дії, відмінний від такого інших класів холестеринознижувальних препаратів (наприклад, статинів, секвестрантів жовчних кислот (смоли), кислотних похідних фібратів та рослинних станолів). Молекулярною мішенню езетимібу є переносник стеролів Niemann-Pick Cl-Like 1 (NPC1L1), що відповідає за всмоктування холестерину та фітостеролів у кишечнику.
Езетиміб локалізується на щітковій облямівці тонкої кишки і пригнічує абсорбцію холестерину, зменшуючи доставку інтестинального холестерину у печінку; статини знижують синтез холестерину у печінці, і разом ці механізми забезпечують додаткове зниження холестерину. Після двотижневого клінічного застосування у 18 пацієнтів з гіперхолестеринемією езетиміб на 54 % знижував абсорбцію холестерину порівняно з плацебо.
Фармакодинамічні ефекти
Була проведена серія доклінічних досліджень для визначення селективності езетимібу щодо пригнічення абсорбції холестерину. Езетиміб пригнічував абсорбцію [14C]-холестерину без впливу на абсорбцію тригліцеридів, жирних кислот, жовчних кислот, прогестерону, етинілестрадіолу або жиророзчинних вітамінів А і D.
Епідеміологічні дослідження встановили, що серцево-судинна захворюваність та летальність змінюються прямо пропорційно до рівня загального холестерину (ХС) та ХС ЛПНЩ (ліпопротеїнів низької щільності) і обернено пропорційно до рівня ХС ЛПВЩ (ліпопротеїнів високої щільності).
Застосування езетимібу зі статином ефективно знижує ризик серцево-судинних ускладнень у пацієнтів з ішемічною хворобою серця та гострим коронарним синдромом (ГКС) в анамнезі.
Клінічна ефективність та безпека
У контрольованих клінічних дослідженнях езетиміб як монотерапія, а також із супутнім застосуванням зі статином значно знижував загальний холестерин (загальний ХС), холестерин ліпопротеїнів низької щільності (ХС ЛПНЩ), аполіпопротеїн В (Апо-В) та тригліцериди (ТГ) і підвищував холестерин ліпопротеїнів високої щільності (ХС ЛПВЩ) у пацієнтів із гіперхолестеринемією.
Первинна гіперхолестеринемія
У подвійно сліпому плацебоконтрольованому 8-тижневому дослідженні 769 пацієнтів з гіперхолестеринемією, які вже отримували монотерапію статинами і у яких ХС ЛПНЩ не відповідав цільовому рівню відповідно до Національної освітньої програми щодо холестерину (NCEP) [США] (від 2,6 до 4,1 ммоль/л [від 100 до 160 мг/дл], залежно від базових характеристик), були рандомізовані для прийому езетимібу 10 мг або плацебо, додатково до поточної терапії статинами.
Серед пацієнтів, які отримували статини і у яких ХС ЛПНЩ не відповідав цільовому рівню на початковому етапі (~ 82 %), значно більше пацієнтів, рандомізованих для прийому езетимібу, досягнули цільового рівня ХС ЛПНЩ в кінцевій точці дослідження порівняно з пацієнтами, рандомізованими для прийому плацебо: відповідно 72 % та 19 %. Відповідні зниження ХС ЛПНЩ суттєво відрізнялися (25 % та 4 % при застосуванні езетимібу порівняно з плацебо відповідно). Крім того, езетиміб, доданий до поточної терапії статинами, суттєво знизив загальний ХС, Апо-В, ТГ і підвищив ХС ЛПНЩ порівняно з плацебо. Езетиміб і плацебо, додані до терапії статинами, зменшили середній рівень С-реактивного білка на 10 % і 0 % від початкового рівня відповідно.
У двох подвійно сліпих рандомізованих плацебоконтрольованих 12-тижневих дослідженнях з участю 1719 пацієнтів з первинною гіперхолестеринемією езетиміб у дозі 10 мг суттєво знизив загальний ХС (13 %), ЛПНЩ (19 %), Апо-В (14 %) і ТГ (8 %) і підвищив ХС ЛПВЩ (3 %) порівняно з плацебо. Крім того, езетиміб не впливав на концентрацію жиророзчинних вітамінів А, D та Е, не впливав на протромбіновий час і, як інші ліпідознижувальні препарати, не заважав продукуванню адренокортикального стероїдного гормону.
У багатоцентровому подвійно сліпому контрольованому клінічному дослідженні (ENHANCE) 720 пацієнтів із гетерозиготною сімейною гіперхолестеринемією були рандомізовані для прийому езетимібу 10 мг у комбінації з симвастатином 80 мг (n = 357) або симвастатину 80 мг (n = 363) протягом 2 років. Основною метою дослідження було вивчення ефекту комбінованої терапії езетимібом/симвастатином на товщину інтима-медіа (ТІМ) сонних артерій порівняно з монотерапією симвастатином. Вплив цього сурогатного маркера на серцево-судинну захворюваність та летальність все ще не продемонстровано. Первинна кінцева точка, зміна середньої ТІМ всіх шести сегментів сонних артерій, не відрізнялася значною мірою (р = 0,29) між двома групами лікування, як було визначено за допомогою ультразвукового дослідження у В-режимі. При застосуванні езетимібу 10 мг у комбінації з симвастатином 80 мг або тільки симвастатину 80 мг товщина інтима-медіа збільшилася відповідно на 0,0111 мм і 0,0058 мм протягом дворічного дослідження (початкова товщина ТІМ сонних артерій — 0,68 мм та 0,69 мм відповідно).
Езетиміб 10 мг у комбінації з симвастатином 80 мг зменшив рівень ХС ЛПНЩ, загальний ХС, Апо-B і ТГ значно більше, ніж симвастатин 80 мг. Процентне збільшення ХС ЛПВЩ було подібним для двох груп лікування. Побічні реакції, про які повідомляли при застосуванні езетимібу 10 мг у комбінації з симвастатином 80 мг, відповідали його відомому профілю безпеки.
Діти
У багатоцентровому подвійно сліпому контрольованому дослідженні 138 пацієнтів (59 хлопчиків та 79 дівчат) віком від 6 до 10 років (середній вік 8,3 року) із гетерозиготною сімейною або несімейною гіперхолестеринемією (ГеСГ) з початковими рівнями ХС ЛПНЩ між 3,74 і 9,92 ммоль/л були рандомізовані для прийому езетимібу 10 мг або плацебо протягом 12 тижнів.
На 12-му тижні езетиміб суттєво знизив загальний ХС (–21 % проти 0 %), ХС ЛПНЩ (–28 % проти –1 %), Aпо-B (–22 % проти –1 %), ХС ЛПВЩ (–26 % проти 0 %) порівняно з плацебо. Результати у двох групах лікування були подібними для ТГ та ХС ЛПВЩ (–6 % проти +8 % та +2 % проти +1 % відповідно).
У багатоцентровому подвійно сліпому контрольованому дослідженні 142 хлопчики (стадія Таннера II або вище) та 106 дівчаток після менархе віком від 10 до 17 років (середній вік 14,2 року) із гетерозиготною сімейною гіперхолестеринемією (ГеСГ) з початковими рівнями ХС ЛПНЩ між 4,1 та 10,4 ммоль/л були рандомізовані для прийому езетимібу 10 мг супутньо з симвастатином (10, 20 або 40 мг) або тільки симвастатину (10, 20 або 40 мг) протягом
6 тижнів, езетимібу і 40 мг симвастатину або тільки 40 мг симвастатину протягом наступних 27 тижнів з подальшим відкритим супутнім прийомом езетимібу та симвастатину (10 мг, 20 мг або 40 мг) протягом 20 тижнів.
На 6-му тижні езетиміб із супутнім застосуванням симвастатину (всі дози) суттєво зменшив загальний ХС (38 % проти 26 %), ХС ЛПНЩ (49 % проти 34 %), Aпо-B (39 % проти 27 %), ХС ЛПВЩ (47 % проти 33 %) порівняно з монотерапією симвастатином (всі дози). Результати у двох групах лікування були подібними для ТГ та ХС ЛПВЩ (–17 % проти –12 % та +7 % проти +6 % відповідно). На 33-му тижні результати були порівнянними з результатами на 6-му тижні, і значно більше пацієнтів, які отримували езетиміб та 40 мг симвастатину (62 %), досягли цільового рівня відповідно до NCEP (< 2,8 ммоль/л [110 мг/дл]) для ХС ЛПНЩ порівняно з тими, хто отримував 40 мг симвастатину (25 %). На 53-му тижні, наприкінці відкритого додаткового дослідження, вплив на ліпідні параметри зберігався.
Безпека та ефективність езетимібу, що призначали у комбінації з симвастатином у дозі більше 40 мг на добу, дітям віком від 10 до 17 років, не вивчалися. Безпеку та ефективність езетимібу, що призначали у комбінації з симвастатином, у дітей віком до 10 років не вивчали. Довгострокова ефективність терапії езетимібом у пацієнтів віком до 17 років для зниження захворюваності та летальності у зрілому віці не досліджувалася.
Профілактика серцево-судинних ускладнень
У міжнародному дослідженні ефективності комбінованого препарату симвастатину/езетимібу (IMPROVE-IT — багатоцентрове рандомізоване подвійно сліпе дослідження з активним контролем) взяли участь 18144 пацієнти, які були включені протягом 10 днів після госпіталізації через гострий коронарний синдром (ГКС, гострий інфаркт міокарда [ІМ] або нестабільна стенокардія [НС]). На момент прояву ГКС ХС ЛПНЩ пацієнтів був ≤ 125 мг/дл (≤ 3,2 ммоль/л), якщо вони не отримували ліпідознижувальну терапію, або ≤ 100 мг/дл (≤ 2,6 ммоль/л), якщо вони отримували ліпідознижувальну терапію. Усі пацієнти були рандомізовані у співвідношенні 1:1 для прийому або езетимібу/симвастатину 10/40 мг (n = 9067) або симвастатину 40 мг (n = 9077) та знаходилися під наглядом у середньому 6 років.
Середній вік пацієнтів становив 63,6 року; 76 % були чоловічої статі; 84 % були європейського походження; 27 % мали діабет. Середній показник ХС ЛПНЩ на момент дослідження був 80 мг/дл (2,1 ммоль/л) у пацієнтів, що отримували ліпідознижувальну терапію (n = 6390), та 101 мг/дл (2,6 ммоль/л) у тих пацієнтів, які не отримували попередню ліпідознижувальну терапію (n = 11 594). До госпіталізації у зв’язку з ГКС 34 % пацієнтів отримували терапію статинами. Протягом 1 року середній показник ХС ЛПНЩ у пацієнтів, які продовжували терапію, становив 53,2 мг/дл (1,4 ммоль/л) у групі езетимібу/симвастатину і 69,9 мг/дл (1,8 ммоль/л) у групі монотерапії симвастатином. Значення ліпідів загалом були отримані для пацієнтів, які залишилися в досліджуваній терапії.
Первинною кінцевою точкою була комбінація, що включала летальний наслідок з причини серцево-судинної патології, гострий коронарний синдром (ГКС, що визначався як нелетальний інфаркт міокарда, задокументована нестабільна стенокардія, яка потребувала госпіталізації, або будь-яка процедура коронарної реваскуляризації, яка проходила як мінімум через 30 днів після призначення рандомізованого лікування) і нелетальний інсульт. Дослідження показали, що лікування езетимібом при додаванні до симвастатину забезпечує поступову перевагу у зменшенні первинної комбінованої кінцевої точки летального наслідку з причини серцево-судинної патології, ГКС та нелетального інсульту порівняно з симвастатином (відносний ризик знижується на 6,4 %, р = 0,016). Первинна кінцева точка спостерігалася у 2572 з 9067 пацієнтів (7-річний показник на основі методу Каплана — Меєра [KM] 32,72 %) в групі езетимібу/симвастатину та 2742 з 9077 пацієнтів (7-річний показник на основі методу КМ 34,67 %) у групі симвастатину (див. рис.1 і таблицю 1). Очікується, що ця додаткова перевага буде схожою з одночасним введенням інших статинів, які показали свою ефективність у зменшенні ризику серцево-судинних ускладнень. Загальна летальність у цій групі високого ризику не змінилася (див. таблицю 1).
Загалом відзначалося зменшення ризику всіх типів інсультів; проте спостерігалося незначне збільшення ризику геморагічного інсульту в групі застосування езетимібу/симвастатину порівняно з симвастатином (див. таблицю 1). Ризик геморагічного інсульту при застосуванні езетимібу разом із високоактивними статинами у довгострокових дослідженнях результатів не був оцінений.
Лікувальний ефект езетимібу/симвастатину в цілому відповідав загальним результатам у багатьох підгрупах, включаючи підгрупи відповідно до статі, віку, раси, наявності цукрового діабету в анамнезі, початкових рівнів ліпідів, попередньої терапії статинами, наявності попереднього інсульту та гіпертонії.
Рис. 1. Вплив езетимібу/симвастатину на первинну комбіновану кінцеву точку, що включає летальний наслідок з причини серцево-судинної патології, гострий коронарний синдром та нелетальний інсульт.
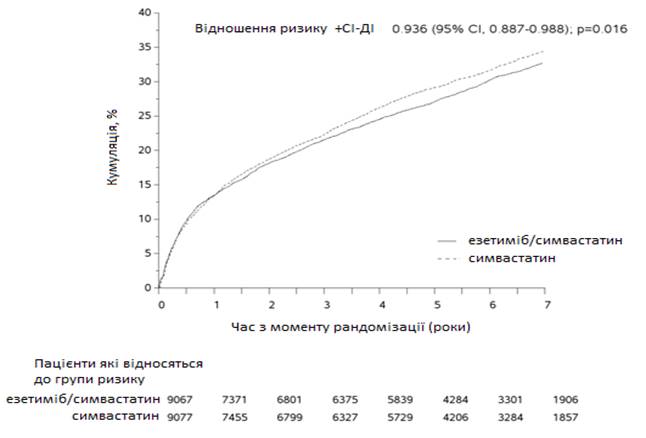
Основні серцево-судинні ускладнення за групами лікування у всіх рандомізованих пацієнтів у дослідженні IMPROVE-IT.
Таблиця 1.
|
Результат |
Езетиміб/симвастатин 10/40 мгa (n = 9067) |
Симвастатин (n = 9077) |
Відношення ризиків (95 % ДІ) |
p-значення |
||
|
n |
КМ % c |
n |
КМ % c |
|||
|
Первинна комбінована кінцева точка |
||||||
|
Летальний наслідок з причини серцево-судинної патології, гострий коронарний синдром і нелетальний інсульт |
2572 |
32,72 % |
2742 |
34,67 % |
0,936 (0,887, 0,988) |
0,016 |
|
Вторинні комбіновані кінцеві точки |
||||||
|
Летальний наслідок через ішемічну хворобу серця (ІХС), нелетальний інфаркт міокарда (ІМ), термінова коронарна реваскуляризація після 30 днів |
1322 |
17,52 % |
1448 |
18,88 % |
0,912 (0,847, 0,983) |
0,016 |
|
ГКС, нелетальний інсульт, летальний наслідок (всі причини) |
3089 |
38,65 % |
3246 |
40,25 % |
0,948 (0,903, 0,996) |
0,035 |
|
Летальний наслідок з причини серцево-судинної патології, нелетальний ІМ, нестабільна стенокардія, яка потребувала госпіталізації, будь-яка реваскуляризація, нелетальний інсульт |
2716 |
34,49 % |
2869 |
36,20 % |
0,945 (0,897, 0,996) |
0,035 |
|
Компоненти первинної комбінованої кінцевої точки та відібрані кінцеві точки ефективності (перші випадки зазначеного ускладнення у будь-який час) |
||||||
|
Летальний наслідок з причини серцево-судинної патології |
537 |
6,89 % |
538 |
6,84 % |
1,000 (0,887, 1,127) |
0,997 |
|
Гострий коронарний синдром: |
||||||
|
Нелетальний ІМ |
945 |
12,77 % |
1083 |
14,41 % |
0,871 (0,798, 0,950) |
0,002 |
|
Нестабільна стенокардія, яка потребувала госпіталізації |
156 |
2,06 % |
148 |
1,92 % |
1,059 (0,846, 1,326) |
0,618 |
|
Термінова коронарна реваскуляризація після 30 днів |
1690 |
21,84 % |
1793 |
23,36 % |
0,947 (0,886, 1,012) |
0,107 |
|
Нелетальний інсульт |
245 |
3,49 % |
305 |
4,24 % |
0,802 (0,678, 0,949) |
0,010 |
|
Усі ІМ (летальний та нелетальний) |
977 |
13,13 % |
1118 |
14,82 % |
0,872 (0,800, 0,950) |
0,002 |
|
Усі інсульти (летальний та нелетальний) |
296 |
4,16 % |
345 |
4,77 % |
0,857 (0,734, 1,001) |
0,052 |
|
Негеморагічний інсультd |
242 |
3,48 % |
305 |
4,23 % |
0,793 (0,670, 0,939) |
0,007 |
|
Геморагічний інсульт |
59 |
0,77 % |
43 |
0,59 % |
1,377 (0,930, 2,040) |
0,110 |
|
Летальний наслідок з будь-якої причини |
1215 |
15,36 % |
1231 |
15,28 % |
0,989 (0,914, 1,070) |
0,782 |
a 6 % пацієнтам доза езетимібу/симвастатину була підвищена до 10/80 мг.
a 27 % пацієнтам доза симвастатину була підвищена до 80 мг.
c 7-річний показник на основі методу Каплана — Меєра.
d Включає ішемічний інсульт або інсульт невизначеного типу.
Профілактика основних судинних ускладнень при хронічному захворюванні нирок
SHARP (дослідження щодо захисту серця та нирок) — це багатонаціональне рандомізоване плацебоконтрольоване подвійно сліпе дослідження з участю 9438 пацієнтів із хронічним захворюванням нирок (ХЗН), третина з яких на початковому етапі знаходилися на діалізі. Загалом 4650 пацієнтам було призначено фіксовану дозу комбінації езетимібу 10 мг із симвастатином 20 мг, а 4620 — плацебо; пацієнти знаходилися під наглядом у середньому 4,9 року. Середній вік пацієнтів становив 62 роки; 63 % були чоловічої статі; 72 % були європейського походження; 23 % мали діабет; у тих, хто не отримував діаліз, середня оцінювана швидкість клубочкової фільтрації (ШКФ) становила 26,5 мл / хв / 1,73 м2. Критеріїв введення ліпідів не було. Середній рівень ХС ЛПНЩ на початковому етапі становив 108 мг/дл. Через рік ХС ЛПНЩ знизився на 26 % при монотерапії симвастатином у дозі 20 мг та на 38 % при прийомі езетимібу у дозі 10 мг в комбінації з симвастатином у дозі 20 мг порівняно з плацебо, у тому числі у пацієнтів, які більше не приймали досліджувані ліки.
Первинне порівняння, яке було визначено у протоколі SHARP, — це аналіз відповідно до початково призначеного лікування «основних судинних ускладнень» (ОСУ: нелетальний ІМ або зупинка серця, інсульт або будь-яка процедура реваскуляризації) тільки в тих пацієнтів, які спочатку були рандомізовані в групу езетимібу в комбінації з симвастатином (n = 4193) або в групу плацебо (n = 4191). Вторинні аналізи включали аналогічну комбінацію, проаналізовану для всієї когорти, рандомізованої (на початковому етапі дослідження або через 1 рік) для прийому езетимібу в комбінації з симвастатином (n = 4650) або плацебо (n = 4620), а також компоненти цієї комбінації.
Аналіз первинної кінцевої точки показав, що езетиміб у комбінації із симвастатином значно зменшив ризик розвитку судинних ускладнень (749 пацієнтів з ускладненнями у групі плацебо та 639 у групі езетимібу в поєднанні з групою симвастатину) з відносним зменшенням ризику на 16 % (р = 0,001 ).
Проте цей план дослідження не дозволив визначити окремо внесок монокомпонентного езетимібу в ефективність, щоб значно зменшити ризик виникнення судинних ускладнень у пацієнтів із ХЗН.
Окремі компоненти ОСУ у всіх рандомізованих пацієнтів представлені в таблиці 2. Езетиміб у комбінації із симвастатином значно зменшив ризик інсульту та потреби у реваскуляризації.
Крім того, ця комбінація показала кращі результати у зменшенні ризику нелетального інфаркту міокарда (ІМ) та зупинки серця порівняно з симвастатином окремо.
Основні судинні ускладнення за групами лікування у всіх рандомізованих пацієнтів у дослідженні SHARPa.
Таблиця 2.
|
Результат |
Езетиміб 10 мг у комбінації з симвастатином 20 мг (n = 4650) |
Плацебо (n = 4620) |
Співвідношення ризику (95 % ДІ) |
Р-значення |
|
Основні судинні ускладнення |
701 (15,1 %) |
814 (17,6 %) |
0,85 (0,77 – 0,94) |
0,001 |
|
Нелетальний ІМ |
134 (2,9 %) |
159 (3,4 %) |
0,84 (0,66 – 1,05) |
0,12 |
|
Зупинка серця |
253 (5,4 %) |
272 (5,9 %) |
0,93 (0,78 – 1,10) |
0,38 |
|
Будь-який інсульт |
171 (3,7 %) |
210 (4,5 %) |
0,81 (0,66 – 0,99) |
0,038 |
|
Негеморагічний інсульт |
131 (2,8 %) |
174 (3,8 %) |
0,75 (0,60 – 0,94) |
0,011 |
|
Геморагічний інсульт |
45 (1,0 %) |
37 (0,8 %) |
1,21 (0,78 – 1,86) |
0,40 |
|
Будь-яка реваскуляризація |
284 (6,1 %) |
352 (7,6 %) |
0,79 (0,68 – 0,93) |
0,004 |
|
Основні атеросклеротичні ускладнення (ОАУ)b |
526 (11,3 %) |
619 (13,4 %) |
0,83 (0,74 – 0,94) |
0,002 |
a Аналіз відповідно до початково призначеного лікування для всіх пацієнтів SHARP, рандомізованих для прийому езетимібу в комбінації з симвастатином або плацебо як на початковому етапі, так і через 1 рік.
b ОАУ; визначаються як комбінація нелетального інфаркту міокарда, коронарної смерті, негеморагічного інсульту або будь-якої реваскуляризації.
Абсолютне зниження рівня холестерину ЛПНЩ, досягнуте застосуванням езетимібу у комбінації з симвастатином, було нижчим серед пацієнтів з нижчим початковим рівнем ХС ЛПНЩ (< 2,5 ммоль/л) та пацієнтів, які отримували діаліз на початковому етапі, ніж у інших пацієнтів, а відповідне зниження ризику в цих двох групах було меншим.
Гомозиготна сімейна гіперхолестеринемія (ГоСГ)
У подвійно сліпому рандомізованому 12-тижневому дослідженні взяли участь 50 пацієнтів з клінічним та/або генотиповим діагнозом ГоСГ, які отримували аторвастатин або симвастатин (40 мг), із супутнім аферезом ЛПНЩ чи без. Езетиміб супутньо з аторвастатином (40 або 80 мг) чи симвастатином (40 або 80 мг) значно знизив рівень ХС ЛПНЩ (на 15 %) порівняно зі збільшенням дози симвастатину або монотерапією аторвастатином від 40 до 80 мг.
Гомозиготна ситостеролемія (фітостеролемія)
У подвійно сліпому плацебоконтрольованому 8-тижневому дослідженні 37 пацієнтів із гомозиготною ситостеролемією були рандомізовані для прийому езетимібу 10 мг (n = 30) або плацебо (n = 7). Деякі пацієнти отримували інше лікування (наприклад, статини, смоли). Езетиміб значно знизив обидва основні рослинні стероли, ситостерол та кампестерол, відповідно на 21 % та 24 % порівняно з початковим рівнем. Вплив зменшення ситостеролу на захворюваність та летальність у цій популяції невідомий.
Стеноз аорти
SEAS (дослідження застосування симвастатину та езетимібу для лікування стенозу аорти) — це багатоцентрове подвійно сліпе плацебоконтрольоване дослідження середньою тривалістю 4,4 року з участю 1873 пацієнтів із безсимптомним стенозом аорти (СА), підтвердженим результатами Допплер-ехокардіографії, при якій швидкість потоку піка аорти була в межах від 2,5 до 4,0 м/с. Лише ті пацієнти, які, на думку лікарів, не потребували лікування статинами з метою зменшення ризику атеросклеротичних серцево-судинних захворювань, були зараховані у дослідження. Пацієнти були рандомізовані 1:1 для прийому плацебо або супутнього введення езетимібу 10 мг та симвастатину 40 мг щоденно.
Первинною кінцевою точкою була комбінація основних серцево-судинних ускладнень (ОСУ), до яких входив летальний наслідок з причини серцево-судинної патології, заміна аортального клапана (ЗАК), застійна серцева недостатність (ЗСН) внаслідок прогресування СА, нелетальний інфаркт міокарда, шунтування коронарної артерії (ШКА), черезшкірне коронарне втручання (ЧКВ), госпіталізація при нестабільній стенокардії та негеморагічний інсульт. Ключовими вторинними кінцевими точками були комбінації підкласів категорій явищ первинних кінцевих точок.
Порівняно з плацебо, езетиміб/симвастатин 10/40 мг несуттєво знизив ризик ОСУ. Первинний результат спостерігався у 333 пацієнтів (35,3 %) у групі езетимібу/симвастатину та у 355 пацієнтів (38,2 %) у групі плацебо (відношення ризику в групі езетимібу/симвастатину 0,96; 95 % довірчий інтервал (ДІ): від 0,83 до 1,12; р = 0,59). Заміна аортального клапана була проведена у 267 пацієнтів (28,3 %) у групі езетимібу/симвастатину та у 278 пацієнтів (29,9 %) у групі плацебо (відношення ризику 1,00; 95 % ДІ: від 0,84 до 1,18; р = 0,97). Менше пацієнтів мали ішемічні серцево-судинні ускладнення в групі езетимібу/симвастатину (n = 148), ніж у групі плацебо (n = 187) (відношення ризику, 0,78; 95 % ДІ: від 0,63 до 0,97; p = 0,02), головним чином через меншу кількість пацієнтів, які перенесли шунтування коронарної артерії.
Рак спостерігався частіше у групі езетимібу/симвастатину (105 проти 70, р = 0,01). Клінічна значущість цього спостереження є невизначеною, оскільки в дослідженні SHARP більша кількість пацієнтів з будь-яким уперше виявленим раком (438 у групі езетимібу/симвастатину та 439 в групі плацебо) не відрізнялися. Крім того, у дослідженні IMPROVE-IT загальна кількість пацієнтів з будь-якою новою злоякісною пухлиною (853 у групі езетимібу/симвастатину та 863 у групі симвастатину) істотно не відрізнялася, і тому результати дослідження SEAS не можуть бути підтверджені дослідженням SHARP або IMPROVE-IT.
Фармакокінетика
Абсорбція
Після прийому всередину езетиміб швидко всмоктується та активно кон’югує з утворенням фармакологічно активного фенольного глюкуроніду (езетиміб-глюкуронід). Середня максимальна концентрація (Сmах) у плазмі крові досягається через 1–2 години для езетимібу-глюкуроніду і через 4–12 години для езетимібу. Абсолютну біодоступність езетимібу визначити неможливо, оскільки ця сполука нерозчинна у водному середовищі, придатному для ін’єкцій.
Одночасний прийом їжі (з низьким або високим вмістом жиру) не впливає на пероральну біодоступність езетимібу. Езетиміб можна приймати незалежно від прийому їжі.
Розподіл
Езетиміб та езетиміб-глюкоронід зв’язуються з білками плазми крові людини на 99,7 % та 88–92 % відповідно.
Метаболізм
Первинний метаболізм езетимібу відбувається в тонкому кишечнику і печінці шляхом кон’югації з глюкуронідом (реакція II фази) з подальшим виведенням із жовчю. Мінімальний окисний метаболізм (реакція I фази) спостерігався на всіх етапах трансформації. Езетиміб та езетиміб-глюкуронід є основними речовинами, що визначаються у плазмі крові, і становлять приблизно 10–20 % і 80–90 % від загального вмісту препарату у плазмі відповідно. Езетиміб та езетиміб-глюкуронід повільно виводяться з плазми крові в процесі кишково-печінкової рециркуляції. Період напіввиведення езетимібу та езетимібу-глюкуроніду становить приблизно 22 години.
Виведення
Після прийому добровольцями всередину 20 мг 14С-езетимібу у плазмі крові було виявлено приблизно 93 % сумарного езетимібу від загальної радіоактивності плазми. Приблизно 78 % і 11 % прийнятої радіоактивної дози було виведено з калом і сечею відповідно протягом 10 днів. Через 48 годин сліди радіоактивності у плазмі крові не виявлялися.
Особливі групи пацієнтів
Діти
Фармакокінетика езетимібу схожа у дітей віком від 6 років і дорослих. Фармакокінетичних даних щодо застосування дітям віком до 6 років немає. Клінічний досвід у дітей і підлітків включає пацієнтів з ГоСГ, ГеСГ або ситостеролемією.
Пацієнти літнього віку. У пацієнтів літнього віку (понад 65 років) концентрація у плазмі крові загального езетимібу приблизно вдвічі вища, ніж у молодших пацієнтів (18–45 років). Зниження ХС ЛПНЩ і профіль безпеки приблизно однакові у пацієнтів літнього віку і молодих пацієнтів, які приймають езетиміб. Тому для пацієнтів літнього віку немає потреби в корекції дози.
Печінкова недостатність. Після одноразового прийому 10 мг езетимібу значення середньої площі під кривою «концентрація — час» (АUC) для загального езетимібу було в 1,7 раза вищим у пацієнтів із печінковою недостатністю легкого ступеня (5–6 балів за шкалою Чайлда — П’ю), ніж у здорових добровольців. Протягом 14-денного дослідження з багаторазовим прийомом езетимібу (по 10 мг щодня) у пацієнтів із печінковою недостатністю помірного ступеня (7–9 балів за шкалою Чайлда — П’ю) значення AUC для загального езетимібу зростало приблизно в 4 рази в 1-й та 14-й день порівняно з таким показником у здорових добровольців. Пацієнтам із печінковою недостатністю легкого ступеня корекція дози не потрібна. Оскільки ефекти підвищеного вмісту езетимібу у пацієнтів із печінковою недостатністю помірного або тяжкого ступеня (більше 9 балів за шкалою Чайлда — П’ю) невідомі, езетиміб не рекомендований для застосування цій категорії пацієнтів (див. розділ «Особливості застосування»).
Ниркова недостатність. Після одноразового прийому 10 мг езетимібу у пацієнтів із тяжкою нирковою недостатністю (n = 8; кліренс креатиніну ≤ 30 мл / хв / 1,73 м2) середнє значення AUC для загального езетимібу зростало приблизно в 1,5 раза порівняно з таким показником у здорових добровольців (n = 9). Цей результат не вважається клінічно значущим. Для пацієнтів із порушенням функції нирок немає потреби в корекції дози.
В одного пацієнта в цьому дослідженні (який мав нирковий трансплантат і отримував мультитерапію, в тому числі циклоспорин) рівень загального езетимібу був вищий у 12 разів.
Стать. Концентрація у плазмі крові загального езетимібу трохи вища (приблизно 20 %) у жінок, ніж у чоловіків. Зниження рівня ХС ЛПНЩ і профіль безпеки приблизно однакові в чоловіків і жінок, які приймають езетиміб. Тому немає потреби в корекції дози залежно від статі.
Первинна гіперхолестеринемія
Езетиміб у комбінації з інгібітором редуктази гідроксиметилглутарил-коензиму А (ГМГ-КоА) (статин) показаний як допоміжна терапія до дієти для пацієнтів з первинною (гетерозиготною сімейною та несімейною) гіперхолестеринемією, якщо терапії тільки статином недостатньо.
Монотерапія езетимібом показана як допоміжна терапія до дієти для пацієнтів з первинною (гетерозиготною сімейною та несімейною) гіперхолестеринемією, для яких застосування статину є недоцільним або існує його непереносимість.
Профілактика серцево-судинних подій
Езетиміб показаний для зменшення ризику серцево-судинних подій (див. розділ «Фармакодинаміка») у пацієнтів з ішемічною хворобою серця (ІХС) та гострим коронарним синдромом (ГКС) в анамнезі при додаванні до поточної терапії статинами або застосуванні одночасно зі статином.
Гомозиготна сімейна гіперхолестеринемія (ГоСГ)
Езетиміб у комбінації зі статином показаний як допоміжна терапія до дієти для пацієнтів із ГоСГ. Пацієнти можуть також отримувати додаткове лікування (наприклад аферез ЛПНЩ).
Гомозиготна ситостеролемія (фітостеролемія)
Езетиміб показаний як допоміжна терапія до дієти для пацієнтів із гомозиготною сімейною ситостеролемією.
Гіперчутливість до діючої речовини або до будь-якої з допоміжних речовин.
При супутньому прийомі езетимібу з будь-яким статином слід ознайомитися з інструкцією для застосування цього конкретного лікарського засобу.
Сумісна терапія езетимібу з будь-яким статином протипоказана у період вагітності або годування груддю.
Застосування езетимібу в комбінації з будь-яким статином протипоказано пацієнтам із захворюваннями печінки у стадії загострення або нез’ясованими тривалими підвищеннями рівнів сироваткових трансаміназ.
Доклінічні дослідження показали, що езетиміб не індукує ферменти системи цитохрому Р450, що метаболізують препарат. Не спостерігалося жодних клінічно значущих фармакокінетичних взаємодій між езетимібом і препаратами, що метаболізуються ферментами системи цитохрому P450 1A2, 2D6, 2C8, 2C9 та 3A4 або N-ацетилтрансферазою.
У клінічних дослідженнях лікарської взаємодії езетиміб при комбінованій терапії не впливав на фармакокінетику дапсону, декстрометорфану, дигоксину, пероральних контрацептивів (етинілестрадіолу та левоноргестрелу), гліпізиду, толбутаміду або мідазоламу. Циметидин при комбінованій терапії з езетимібом не впливав на біодоступність езетимібу.
Антациди
Одночасний прийом антацидів призводить до зниження ступеня абсорбції езетимібу, але не впливає на його біодоступність. Таке зниження ступеня абсорбції не вважається клінічно значущим.
Холестирамін
При комбінованому застосуванні з холестираміном середнє значення площі під кривою «концентрація — час» (AUC) сумарного езетимібу (езетиміб та езетиміб-глюкуронід) зменшувалося приблизно на 55 %. При додаванні езетимібу до холестираміну поступове зниження холестерину ліпопротеїнів низької щільності (ХС ЛПНЩ) може уповільнитися. Фібрати
Якщо пацієнти отримують фенофібрат та езетиміб, лікарі повинні бути проінформовані про можливий ризик виникнення холелітіазу і захворювання жовчного міхура. При підозрі на холелітіаз у пацієнта, який отримує езетиміб та фенофібрат, показані обстеження жовчного міхура, а таку терапію слід припинити (див. розділ «Побічні реакції»).
Супутнє застосування фенофібрату або гемфіброзилу незначним чином підвищувало загальну концентрацію езетимібу (приблизно у 1,5 і 1,7 раза відповідно).
Одночасне застосування езетимібу з іншими фібратами не вивчали.
Фібрати можуть підвищувати виділення холестерину в жовч, що призводить до холелітіазу. У дослідженнях на тваринах езетиміб іноді підвищував рівень холестерину в жовчі, але не у всіх видів. Літогенний ризик, пов’язаний з терапевтичним застосуванням езетимібу, не можна виключити.
Статини
Жодної клінічно значущої фармакокінетичної взаємодії не було виявлено при комбінованому прийомі езетимібу з аторвастатином, симвастатином, правастатином, ловастатином, флувастатином або розувастатином.
Циклоспорин
У ході дослідження з участю 8 пацієнтів після трансплантації нирки з кліренсом креатиніну > 50 мл/хв при стабільній дозі циклоспорину 1 доза езетимібу 10 мг призводила до підвищення у 3,4 раза (діапазон становить від 2,3 до 7,9 раза) середньої AUC загального езетимібу порівняно з відповідним показником у контрольній популяції здорових пацієнтів, які отримували тільки езетиміб, в іншому дослідженні (n = 17). Ще в одному дослідженні у пацієнта з трансплантованою ниркою і тяжкою нирковою недостатністю, який отримував циклоспорин та багато інших лікарських засобів, зафіксовано 12-разове збільшення експозиції загального езетимібу порівняно з пацієнтами контрольної групи, які отримували тільки езетиміб. Під час перехресного дослідження з двома періодами з участю 12 здорових добровольців щоденне введення 20 мг езетимібу протягом 8 днів з однією дозою 100 мг циклоспорину на 7-й день призвело до збільшення AUC циклоспорину в середньому на 15 % (діапазон від зменшення на 10 % до збільшення на 51 %) порівняно з відповідним показником при введенні однієї дози 100 мг циклоспорину. Контрольоване дослідження впливу одночасного прийому езетимібу на експозицію циклоспорину у пацієнтів із трансплантованою ниркою не проводили. Слід з обережністю розпочинати лікування езетимібом пацієнтам, які приймають циклоспорин. У пацієнтів, які приймають езетиміб та циклоспорин, слід перевіряти концентрації циклоспорину.
Антикоагулянти
Одночасне застосування езетимібу (10 мг 1 раз на добу) не мало значного впливу на біодоступність варфарину та протромбіновий час у ході дослідження з участю 12 здорових дорослих чоловіків. Однак були післяреєстраційні повідомлення про збільшення міжнародного нормалізованого відношення (МНВ) у пацієнтів, яким езетиміб додавали до варфарину або флуїндіону. Якщо езетиміб застосовувати на тлі прийому варфарину, іншого кумаринового антикоагулянту або флуїндіону, слід належним чином перевіряти МНВ.
Діти
Дослідження взаємодії у дітей не проводилися.
При супутньому прийомі езетимібу з будь-яким статином слід ознайомитися з інструкцією для застосування цього конкретного лікарського засобу.
Ферменти печінки
У контрольованих дослідженнях супутньої терапії у пацієнтів, які отримували езетиміб зі статинами, спостерігалися послідовні підвищення рівня трансаміназ (у 3 або більше разів вище за верхню межу норми [ВМН]). При прийомі комбінації езетимібу зі статином слід проводити функціональні печінкові проби на початку терапії та згідно з рекомендаціями стосовно статину.
У міжнародному дослідженні ефективності комбінованого препарату симвастатину/езетимібу (IMPROVE-IT) 18144 пацієнти з ішемічною хворобою серця та випадками ГКС в анамнезі були рандомізовані для прийому езетимібу/симвастатину 10/40 мг щоденно (n = 9067) або симвастатину 40 мг щоденно (n = 9077). Протягом середнього періоду спостереження 6 років частота послідовного підвищення трансаміназ (у 3 або більше разів вище за ВМН) становила 2,5 % у групі прийому езетимібу/симвастатину та 2,3 % у групі симвастатину.
У контрольованому клінічному дослідженні, у якому понад 9000 пацієнтів із хронічним захворюванням нирок були рандомізовані для прийому езетимібу 10 мг у комбінації із симвастатином 20 мг щоденно (n = 4650) або плацебо (n = 4620) (середній період спостереження 4,9 року), частота послідовного підвищення трансаміназ (у 3 або більше разів вище за ВМН) становила 0,7 % у групі прийому езетимібу в комбінації з симвастатином і 0,6 % у групі плацебо.
Скелетні м’язи
У післяреєстраційному періоді повідомляли про випадки міопатії та рабдоміолізу при застосуванні езетимібу. Більшість пацієнтів, у яких розвинувся рабдоміоліз, приймали статин одночасно з езетимібом. Однак про випадки рабдоміолізу повідомляли дуже рідко при монотерапії езетимібом і у разі додавання езетимібу до інших засобів, з якими пов’язаний підвищений ризик виникнення рабдоміолізу. При підозрі на міопатію, що проявляється симптомами з боку м’язів або підвищенням рівня креатинкінази більше ніж у 10 разів від верхньої межі норми, слід негайно припинити прийом езетимібу, будь-якого статину та будь-якого з інших препаратів, які пацієнт приймає одночасно. Усіх пацієнтів, які починають лікування езетимібом, потрібно проінформувати про ризик виникнення міопатії та необхідність терміново повідомляти про появу болю в м’язах, млявості або слабкості незрозумілої етіології.
У дослідженні IMPROVE-IT 18144 пацієнти з ішемічною хворобою серця та випадками ГКС в анамнезі були рандомізовані для прийому езетимібу/симвастатину 10/40 мг щоденно (n = 9067) або симвастатину 40 мг щоденно (n = 9077). Протягом середнього періоду спостереження 6 років частота міопатії становила 0,2 % у групі прийому езетимібу/симвастатину та 0,1 % у групі симвастатину, де міопатія визначалася як м’язова слабкість або біль незрозумілої етіології, коли рівень креатинкінази в сироватці крові був щонайменше у 10 разів вище за ВМН або коли за результатами двох послідовних вимірювань рівень креатинкінази перевищував ВМН у 5 разів і більше, але менше ніж у 10 разів. Частота рабдоміолізу становила 0,1 % у групі прийому езетимібу/симвастатину та 0,2 % у групі симвастатину, де рабдоміоліз визначався як м’язова слабкість або біль незрозумілої етіології, коли рівень креатинкінази в сироватці крові був щонайменше у 10 разів вище за ВМН з ознаками пошкодження нирок або коли за результатами двох послідовних вимірювань рівень креатинкінази перевищував ВМН у 5 разів і більше, але менше ніж у 10 разів, з ознаками пошкодження нирок чи рівнем креатинкінази ≥ 10000 МО/л без ознак пошкодження нирок.
У клінічному дослідженні, у якому понад 9000 пацієнтів із хронічним захворюванням нирок були рандомізовані для прийому езетимібу 10 мг в комбінації із симвастатином 20 мг щоденно (n = 4650) або плацебо (n = 4620) (середній період спостереження 4,9 року), частота міопатії/рабдоміолізу становила 0,2 % у групі прийому езетимібу в комбінації з симвастатином і 0,1 % у групі плацебо.
Печінкова недостатність
Оскільки ефекти підвищеного вмісту езетимібу у пацієнтів із печінковою недостатністю помірного та тяжкого ступеня невідомі, езетиміб не рекомендований для застосування цій категорії пацієнтів.
Діти
Ефективність та безпеку езетимібу у пацієнтів віком від 6 до 10 років із гетерозиготною сімейною або несімейною гіперхолестеринемією оцінювали у плацебоконтрольованому клінічному дослідженні протягом 12 тижнів. Вплив езетимібу протягом періоду лікування більше 12 тижнів у цій віковій групі не вивчали.
Вплив езетимібу не вивчали у пацієнтів віком до 6 років.
Ефективність та безпека езетимібу, що призначали у комбінації із симвастатином пацієнтам віком від 10 до 17 років із гетерозиготною сімейною гіперхолестеринемією, оцінювалися в контрольованому клінічному дослідженні з участю хлопчиків (стадія Таннера II або вище) та дівчаток (не раніше ніж через рік після менархе).
Під час цього обмеженого контрольованого дослідження взагалі не спостерігалося жодного помітного впливу на ріст і статеве дозрівання хлопчиків та дівчаток підліткового віку і жодної дії на тривалість менструального циклу в дівчаток. Однак дію езетимібу на ріст та статеве дозрівання упродовж періоду лікування більше 33 тижнів не вивчали.
Безпеку та ефективність езетимібу, що призначали у комбінації із симвастатином у дозі більше 40 мг на добу, у дітей віком від 10 до 17 років не вивчали.
Безпеку та ефективність езетимібу, що призначали у комбінації із симвастатином, у дітей віком до 10 років не вивчали.
Довгострокову ефективність терапії езетимібом у пацієнтів віком до 17 років для зниження захворюваності та летальності у зрілому віці не досліджували.
Фібрати
Безпека та ефективність езетимібу при застосуванні з фібратами не встановлена.
При підозрі на холелітіаз у пацієнта, який отримує езетиміб та фенофібрат, показані обстеження жовчного міхура, а таку терапію слід припинити.
Циклоспорин
Слід з обережністю розпочинати лікування езетимібом пацієнтам, які приймають циклоспорин. У пацієнтів, які приймають езетиміб та циклоспорин, слід перевіряти концентрації циклоспорину.
Антикоагулянти
Якщо езетиміб застосовувати на тлі прийому варфарину, іншого кумаринового антикоагулянту або флуїндіону, слід належним чином перевіряти міжнародне нормалізоване відношення (МНВ).
Вплив допоміжних речовин
Пацієнтам з рідкісними спадковими проблемами непереносності галактози, дефіцитом лактази або глюкозо-галактозною мальабсорбцією не слід приймати цей лікарський засіб.
Ліпобон містить менше 1 ммоль (23 мг) натрію на таблетку, тобто практично вільний натрію.
Застосування у період вагітності або годування груддю
Терапія езетимібом у комбінації зі статином протипоказана у період вагітності або годування груддю (див. розділ «Протипоказання»); слід ознайомитися з інструкцією для застосування цього статину.
Вагітність
Езетиміб слід призначати вагітним жінкам, тільки якщо це явно необхідно. Клінічні дані про застосування езетимібу у період вагітності відсутні. Дослідження на тваринах із використанням езетимібу як монотерапії показали відсутність прямого або непрямого шкідливого впливу на вагітність, ембріофетальний розвиток, пологи або постнатальний період розвитку.
Годування груддю
Езетиміб не слід застосовувати у період годування груддю. Дослідження на щурах показали, що езетиміб проникає молоко лактуючих тварин. Невідомо, чи проникає езетиміб у грудне молоко людини.
Фертильність
Клінічні дані про вплив езетимібу на фертильність людини відсутні. Езетиміб не показав жодного впливу.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Досліджень щодо впливу на здатність керувати автомобілем та іншими механічними засобами не проводили. Але слід враховувати, що повідомляли про запаморочення при керуванні автомобілем та іншими механізмами.
Дозування
Протягом усього курсу лікування езетимібом пацієнт повинен дотримуватися стандартної ліпідознижувальної дієти.
Лікарський засіб застосовувати перорально. Рекомендована доза езетимібу становить 10 мг (1 таблетка) на добу. Езетиміб можна приймати у будь-який час доби незалежно від прийому їжі.
При додаванні езетимібу до статину слід продовжувати прийом початкової дози цього статину або більш високої дози статину, яка вже була призначена. У такому випадку слід ознайомитись з інструкцією для медичного застосування цього статину.
Застосування пацієнтам з ішемічною хворобою серця та випадками ГКС в анамнезі
Для поступового зниження ризику виникнення серцево-судинних ускладнень пацієнтам з ішемічною хворобою серця та випадками ГКС в анамнезі езетиміб (10 мг) можна застосовувати у комбінації зі статином з підтвердженим сприятливим впливом на серцево-судинну систему.
Супутній прийом із секвестрантами жовчних кислот
Прийом езетимібу слід проводити не пізніше ніж за 2 години до або не раніше ніж через 4 години після введення секвестранту жовчної кислоти.
Пацієнти літнього віку
Пацієнти літнього віку не потребують корекції дози.
Печінкова недостатність
Для пацієнтів із печінковою недостатністю легкого ступеня (5–6 балів за шкалою Чайлда — П’ю) корекція дози не потрібна. Лікування езетимібом не рекомендовано пацієнтам із помірним (7–9 балів за шкалою Чайлда — П’ю) або тяжким (більше 9 балів за шкалою Чайлда — П’ю) порушенням функції печінки.
Ниркова недостатність
Пацієнти з нирковою недостатністю не потребують корекції дози.
Діти
На початку лікування необхідний нагляд спеціаліста.
Діти віком від 6 років: безпека та ефективність застосування езетимібу дітям віком від 6 до 17 років не встановлена. Наявні на цей час дані наводяться у розділах «Фармакологічні властивості», «Особливості застосування», «Побічні реакції», але рекомендації щодо дозування неможливі.
При застосуванні езетимібу зі статином слід ознайомитися з інструкцією для застосування щодо дозування для дітей.
Діти віком до 6 років: безпека та ефективність застосування езетимібу дітям віком до 6 років не встановлена. Інформація відсутня.
У дослідженнях застосування езетимібу 50 мг/добу 15 здоровим учасникам протягом 14 днів або 40 мг/добу 18 пацієнтам з первинною гіперхолестеринемією протягом до 56 днів, як правило, добре переносилося. У тварин токсичність не спостерігалася: після одноразових пероральних доз 5000 мг/кг езетимібу — у щурів та мишей і 3000 мг/кг — у собак.
Повідомляли про декілька випадків передозування езетимібу; здебільшого передозування не спричинило небажаних явищ. Небажані явища, які спостерігалися, не були серйозними. У разі передозування слід вжити симптоматичних та підтримуючих заходів.
У дослідженнях тривалістю до 112 тижнів езетиміб у дозі 10 мг на добу вводили окремо (2396 пацієнтам) або в комбінації зі статином (11308 пацієнтам) чи фенофібратом (185 пацієнтам). Побічні реакції, як правило, були незначними та короткотривалими. Загальна частота побічних реакцій була подібною у групі прийому езетимібу та групі плацебо. Аналогічно частота припинення лікування через небажані явища була порівнянною у групі прийому езетимібу та групі плацебо.
Езетиміб як монотерапія або в комбінації зі статином
Нижчезазначені побічні реакції спостерігалися у клінічних дослідженнях у пацієнтів, які лікувалися езетимібом (N = 2396), та виникали частіше, ніж у групі плацебо (N = 1159), або спостерігалися у пацієнтів, які лікувалися езетимібом супутньо зі статином (N = 11308), та виникали частіше, ніж у групі прийому тільки статину (N = 9361). Побічні реакції, про які повідомлялося у післяреєстраційний період, виникали при застосуванні езетимібу окремо або супутньо зі статином.
Частота виникнення побічних ефектів класифікується таким чином: дуже часто (≥1/10), часто (≥1/100 до <1/10), нечасто (≥1/1000 до <1/100), рідко (≥1/10000 <1/1000), дуже рідко (<1/10 000) та невідомо (неможливо оцінити за наявними даними).
|
Системи органів |
Побічні реакції |
Частота |
|
З боку обміну речовин та харчування |
Зниження апетиту |
Нечасто |
|
З боку судинної системи |
Припливи; гіпертензія |
Нечасто |
|
З боку дихальної системи, органів грудної клітки та середостіння |
Кашель |
Нечасто |
|
Диспное |
Невідомо |
|
|
З боку шлунково-кишкового тракту |
Абдомінальний біль; діарея; метеоризм |
Часто |
|
Диспепсія; гастроезофагеальний рефлюкс; нудота; сухість у роті; гастрит |
Нечасто |
|
|
панкреатит; закреп |
Невідомо |
|
|
З боку скелетно-м’язової та сполучної тканини |
Міалгія |
Часто |
|
Артралгія; м’язові спазми; біль у шиї; м’язова слабкість; біль у кінцівках |
Нечасто |
|
|
Міопатія/рабдоміоліз |
Невідомо |
|
|
Загальні порушення та реакції у місці введення |
Втома |
Часто |
|
Біль у грудях, біль; астенія; периферичні набряки |
Нечасто |
|
|
Лабораторні дослідження |
Підвищення рівня аланінамінотрансферази (АЛТ) та/або аспартатамінотрансферази (АСТ) |
Часто |
|
Підвищення рівня креатинфосфокінази в сироватці крові; підвищення рівня гамма-глутамілтрансферази; відхилення від норми показників функціональних печінкових проб |
Нечасто |
|
|
З боку системи кровотворення та лімфатичної системи |
Тромбоцитопенія |
Невідомо |
|
З боку імунної системи |
Гіперчутливість, включаючи висипання, кропив’янку, анафілаксію та ангіоневротичний набряк |
Невідомо |
|
Психічні порушення |
Депресія |
Невідомо |
|
З боку нервової системи |
Головний біль |
Часто |
|
Парестезія |
Нечасто |
|
|
Запаморочення |
Невідомо |
|
|
З боку печінки і жовчовивідних шляхів |
Гепатит; холелітіаз; холецистит |
Невідомо |
|
З боку шкіри та підшкірних тканин |
Свербіж; висипання; кропив’янка |
Нечасто |
|
Мультиформна еритема |
Невідомо |
Супутній прийом езетимібу та фенофібрату
Порушення з боку шлунково-кишкового тракту: абдомінальний біль (часто).
У багатоцентровому подвійно сліпому плацебоконтрольованому клінічному дослідженні у пацієнтів зі змішаною гіперліпідемією 625 пацієнтів отримували лікування протягом періоду до 12 тижнів і 576 пацієнтів протягом періоду до 1 року. У цьому дослідженні 172 пацієнти, які отримували лікування езетимібом і фенофібратом, завершили 12-тижневе лікування, а 230 пацієнтів, які отримували лікування езетимібом і фенофібратом (у тому числі 109 пацієнтів, які отримували езетиміб як монотерапію протягом перших 12 тижнів), завершили однорічне лікування. Це дослідження було розроблено не для порівняння груп лікування на предмет виникнення нечастих явищ. Частота виникнення (95 % ДІ) клінічно важливого підвищення (у 3 рази вище за верхню межу норми, послідовного) рівнів сироваткових трасаміназ становила 4,5 % (1,9 і 8,8) та 2,7 % (1,2 і 5,4) при монотерапії фенофібратом та при одночасному застосуванні езетимібу з фенофібратом відповідно, з урахуванням тривалості лікування. Відповідні показники частоти виникнення холецистектомії становили 0,6 % (0,0 і 3,1) та 1,7 % (0,6 і 4,0) при монотерапії фенофібратом та при одночасному застосуванні езетимібу з фенофібратом відповідно, з урахуванням тривалості лікування.
Діти (віком від 6 до 17 років)
Відомо, що під час дослідження з участю дітей віком від 6 до 10 років із гетерозиготною сімейною або несімейною гіперхолестеринемією (n = 138) спостерігалося підвищення рівнів АЛТ і/або АСТ (щонайменше у 3 рази вище за верхню межу норми, послідовне) у 1,1 % (1 пацієнт) пацієнтів, які отримували лікування езетимібом порівняно з 0 % у групі плацебо. Підвищення рівнів креатинфосфокінази (у 10 і більше разів вище за верхню межу норми) не спостерігалося. Про випадки міопатії не повідомляли.
Під час іншого дослідження з участю підлітків (віком від 10 до 17 років) із гетерозиготною сімейною гіперхолестеринемією (N = 248) спостерігалося підвищення рівнів АЛТ та/або АСТ (щонайменше у 3 рази вище за верхню межу норми, послідовне) у 3 % (4 пацієнтів) у групі езетимібу/симвастатину порівняно з 2 % (2 пацієнти) у групі монотерапії симвастатином; ці показники становили відповідно 2 % (2 пацієнти) і 0 % стосовно підвищення рівня креатинфосфокінази (КФК) у 10 і більше разів вище за верхню межу норми. Про випадки міопатії не повідомляли.
У цьому дослідженні не порівнювали рідкісні побічні реакції на препарат.
Пацієнти з ішемічною хворобою серця та випадками ГКС в анамнезі
У дослідженні IMPROVE-IT (див. розділ «Фармакодинаміка») з участю 18144 пацієнтів, які отримували езетиміб/симвастатин 10/40 мг (n = 9067; з яких 6 % пацієнтам було підвищено дози езетимібу/симвастатину до 10/80 мг) або симвастатин 40 мг (n = 9077, з них 27 % було підвищено дози симвастатину до 80 мг), профілі безпеки були однаковими протягом середнього періоду спостереження 6 років. Частота припинення лікування через небажані явища становила 10,6 % у пацієнтів, які отримували езетиміб/симвастатин, і 10,1 % у пацієнтів, які отримували симвастатин. Частота міопатії становила 0,2 % у групі прийому езетимібу/симвастатину та 0,1 % у групі симвастатину, де міопатія визначалася як м’язова слабкість або біль незрозумілої етіології, коли рівень креатинкінази в сироватці крові був щонайменше у 10 разів вище за ВМН або коли за результатами двох послідовних вимірювань рівень креатинкінази перевищував ВМН у 5 разів і більше, але менше ніж у 10 разів. Частота рабдоміолізу становила 0,1 % у групі прийому езетимібу/симвастатину та 0,2 % у групі симвастатину, де рабдоміоліз визначали як м’язову слабкість або біль незрозумілої етіології, коли рівень креатинкінази в сироватці крові був щонайменше у 10 разів вище за ВМН з ознаками пошкодження нирок або коли за результатами двох послідовних вимірювань рівень креатинкінази перевищував ВМН у 5 разів і більше, але менше ніж у 10 разів, з ознаками пошкодження нирок чи рівнем креатинкінази ≥ 10000 МО/л без ознак пошкодження нирок.
Частота послідовного підвищення трансаміназ (щонайменше у 3 рази вище за ВМН) становила 2,5 % у групі прийому езетимібу/симвастатину та 2,3 % у групі симвастатину (див. розділ «Особливості застосування»). Побічні ефекти, пов’язані з жовчним міхуром, відзначалися у 3,1 % та 3,5 % пацієнтів, які отримували езетиміб/симвастатин та симвастатин відповідно. Частота госпіталізації через холецистектомію становила 1,5 % в обох групах лікування. Рак (що визначали як будь-яку нову злоякісну пухлину) був діагностований під час дослідження у 9,4 % та 9,5 % відповідно.
Пацієнти з хронічним захворюванням нирок
У дослідженні SHARP (дослідження щодо захисту серця та нирок) (див. розділ «Фармакодинаміка») з участю понад 9000 пацієнтів, які отримували фіксовану комбінацію езетимібу 10 мг із симвастатином 20 мг щоденно (n = 4650) або плацебо (n = 4620), профілі безпеки були порівнянними протягом середнього періоду спостереження 4,9 року. У цьому дослідженні були зареєстровані лише серйозні побічні реакції та припинення лікування через будь-які побічні реакції. Частота припинення лікування через побічні реакції була порівнянною (10,4 % у пацієнтів, які отримували езетиміб у комбінації із симвастатином, і 9,8 % у пацієнтів, які отримували плацебо). Частота міопатії/рабдоміолізу становила 0,2 % у пацієнтів, які отримували езетиміб у комбінації з симвастатином, і 0,1 % у пацієнтів, які отримували плацебо. Послідовне підвищення рівнів трансаміназ (у 3 рази вище за ВМН) спостерігалося у 0,7 % пацієнтів, які отримували езетиміб у комбінації з симвастатином порівняно з 0,6 % пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»). У цьому дослідженні не було статистично достовірного збільшення частоти попередньо визначених побічних реакцій, включаючи рак (9,4 % при застосуванні езетимібу в комбінації з симвастатином, 9,5 % у групі плацебо), гепатит, холецистектомію, ускладнення жовчнокам’яної хвороби або панкреатиту.
Дані лабораторних досліджень
Під час контрольованих клінічних досліджень монотерапії клінічно значуще підвищення рівнів сироваткових трансаміназ (АЛТ та/або АСТ щонайменше у 3 рази вище за ВМН) було подібним у групі прийому езетимібу (0,5 %) та у групі плацебо (0,3 %). У дослідженнях комбінованої терапії частота появи становила 1,3 % у пацієнтів, які супутньо приймали езетиміб та статин, і 0,4 % у пацієнтів, які приймали тільки статин. Подібне підвищення зазвичай було безсимптомне, не пов’язане з холестазом, а показники поверталися до початкового рівня після відміни терапії або при продовженні лікування.
У клінічних дослідженнях спостерігалося підвищення КФК у 10 або більше разів вище за ВМН у 4 із 1674 пацієнтів (0,2 %), які приймали тільки езетиміб, порівняно з 1 з 786 пацієнтів (0,1 %), які приймали плацебо, та у 1 з 917 пацієнтів (0,1 %), які приймали езетиміб супутньо зі статином, порівняно з 4 з 929 пацієнтів (0,4 %), які приймали тільки статин. Не було переваги у появі міопатії або рабдоміолізу при прийомі езетимібу порівняно з відповідною групою контролю (плацебо або тільки статин).
Звітування про підозрювані побічні реакції
Звітування про підозрювані побічні реакції після реєстрації лікарського засобу має велике значення. Це дає змогу проводити безперервне спостереження співвідношення між користю і ризиками, пов’язаними із застосуванням лікарського засобу. Медичним та фармацевтичним працівникам, а також пацієнтам або їхнім законним представникам слід повідомляти про усі випадки підозрюваних побічних реакцій та відсутності ефективності лікарського засобу через Автоматизовану інформаційну систему з фармаконагляду за посиланням: https://aisf.dec.gov.ua.
3 роки.
Зберігати при температурі не вище 30 °С. Зберігати в оригінальній упаковці у захищеному від вологи місці.
По 10 таблеток в блістері; по 3 або по 6, або по 9 блістерів в картонній пачці.
За рецептом.
ЗАТ Фармацевтичний завод ЕГІС.
Місцезнаходження виробника та адреса місця провадження його діяльності.
1165, м. Будапешт, вул. Бекеньфелді, 118-120, Угорщина.
Езетиміб: 10 мг/таблетка
Популярні питання
Ціна на Ліпобон таблетки по 10 мг №30 (3 блістери х 10 таблеток) стартує від 101.28 ₴ - блістер / 10 шт.
Протипоказано. Детальніше необхідно проконсультуватися з вашим лікарем.
Країна виробник у Ліпобон таблетки по 10 мг №30 (3 блістери х 10 таблеток) - Угорщина.
Основною діючою речовиною у Ліпобон таблетки по 10 мг №30 (3 блістери х 10 таблеток) є Езетиміб.

Переноситься організмом чудово, показники ліпідограми після початку прийому покращилися.